GATKによる変異検出のためのロードマップ [GATK解説シリーズのまとめ記事]
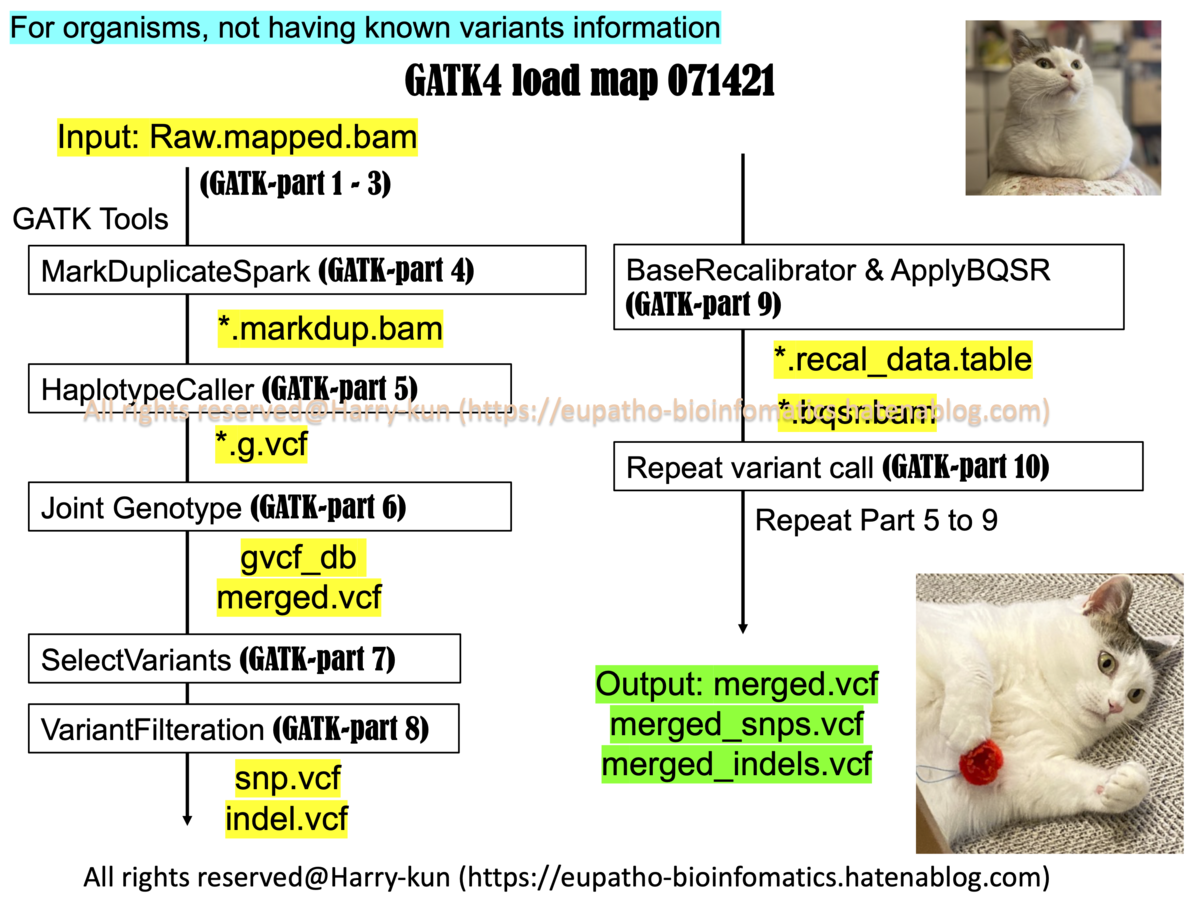
GATKの使い方 BAMファイルからVCF出力までのロードマップ
GATK4.2の使い方について、ロードマップを作成しました。
各partに対応した作業内容について、1つずつ記事にしています。
ちなみに、ブログ主の研究対象がハプロイドの病原体なので、とりあえず1倍体の生物を対象にしています。
いつになるかわかりませんが、ヒト(2倍体ゲノム)を使った内容も勉強して紹介したいと考えています。
新しい関連記事を投稿するごとに順次内容を更新しますので、本ブログでGATKに関する記事を検索したい方はこのページをブックマークされておくと便利に使っていただけると思います。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog
GATK解説シリーズ パート1 GATKの導入
GATKはBroad研究所が提供する、変異の検出に特化したゲノム解析ツールキットです。非常に多機能でゲノム解析分野で業界標準として広く使われていますが、操作難易度が高く、初心者は必ず躓くポイントになっています。GATKはもともとヒトゲノム解析用に開発されたものですが、現在ではあらゆる生物のゲノムデータを扱えるように進化しています。その範囲は、体細胞・生殖細胞のショートバリアント/インデル検出、コピー数(CNV)や構造変異(SV)への解析に及びます。また、GATKには、WGSの処理や品質管理などの関連作業を行うための多くのユーティリティが含まれており、頻繁に使われるPicardも搭載されています。また、GATKの読み方は(ジー・エー・ティー・ケイ)です。公式HPはこちら
eupatho-bioinfomatics.hatenablog.com
GATK解説シリーズ パート2 使用するWGSデータの収集
今後の解説で例として使用するマラリア原虫のWGSデータを収集します。ここでは。fasterq-dumpを使ったダウンロードの方法を紹介します。
この後は、GWASで使用する変異データの取得までを順番に紹介してきます。
eupatho-bioinfomatics.hatenablog.com
GATK解説シリーズ パート3 今後のGATK解析で使用するWGSデータのマッピング
前回の記事で収集したマラリア原虫のWGSデータを使ってマッピングを行い、BAM形式のファイルを生成します。
本記事では、トリミングからマッピングまでの工程をコンパクトに記載しているので、実際にご自身のデータを使って再現される場合には、この記事のスクリプトを置き替える形で使ってもらえると便利だと思います。
eupatho-bioinfomatics.hatenablog.com
GATK解説シリーズ パート4 GATK MarkDuplicatesの使い方
- 前回の記事で得たBAM形式ファイルを使って、GATK MarkDuplicates/MarkDuplicateSparkにより重複したリードにタグを付けます。
eupatho-bioinfomatics.hatenablog.com
GATK解説シリーズ パート5 GATK HaplotypeCallerの使い方
GATK HaplotypeCallerを使って、前回の記事で得たBAM形式ファイルから、変異情報の記載されたVCFファイルを出力します。
前編では、GATK HaplotypeCallerについて解説しました。
後編では、GATK HaplotypeCallerを実践します。
eupatho-bioinfomatics.hatenablog.com
eupatho-bioinfomatics.hatenablog.com
番外編 VCFとはなにかを説明します
- これまでに全くVCFファイルに触れたことのない方に向けて、ファイルを構成する要素を詳しく解説します。
- VCFは変異解析に不可欠な要素であり、決して避けては通れません。 初めて見た時の感想は、「うわ、ちんぷんかんぶん・・・」という印象を持つと思いますが、慣れてくると意外と単純な要素で構成されていることに気が付きます。VCFファイルの形式を理解しないと、GATKやvcf/bcftoolsを使ったフィルタリングで何をしているか理解できなくなってしまうので、この機会に一緒に勉強していきましょう。
- 二つの記事を読み終わる頃には、きっとVCFファイルに対する苦手意識は無くなっていると思います!
前編では、VCFを構成する要素について順番に解説しました。
後編では各バリアントサイトのレコードに付随する項目やスコアについて説明しました。
eupatho-bioinfomatics.hatenablog.com
eupatho-bioinfomatics.hatenablog.com
GATK解説シリーズ パート6GATK JointGenotypingの使い方
- GATK GenomicsDBimport および GATK GenotypeGVCFs を使ったJoint Genotypingを実施して、複数のvcfファイルをまとめたmerged.vcfファイルを出力します。
eupatho-bioinfomatics.hatenablog.com
GATK解説シリーズ パート7 GATK SelectVariantsの使い方
- GATK SelectVariantsを使って、merged.vcfファイルから、自身の解析条件に適したフィルター条件を設定して、バリアントを選別します。
eupatho-bioinfomatics.hatenablog.com
GATK解説シリーズ パート8 GATK VariantFilterationの使い方
- GATK VariantFiltrationを使って、vcfファイルから低クオリティのバリアントを除外します。
eupatho-bioinfomatics.hatenablog.com
GATK解説シリーズ パート9GATK BaseRecalibratorとApplyBQSRの使い方
- 前回のGATK VariantFilterationで出力された、
merged_snps/indels_filtered.vcfを基にBQSRを行います。
eupatho-bioinfomatics.hatenablog.com
GATK解説シリーズ パート10 GATK BQSR後のVariant Calling
前回のGATK VariantFilterationで出力された、
*.bqsr.bamを基に2回目のVariant Callingを行います。今回行う内容は、GATK解説シリーズ Part 5 からPart 8で行った仕事の繰り返しになります。新しく覚えることは特にありません。
今回得た
merged_snps/indels_filtered.vcfを使って、多様なゲノムワイド解析を行うことができます。
eupatho-bioinfomatics.hatenablog.com
GATK解説シリーズ パート11 GATK FastaAlternateReferanceMakerを使って代替参照配列を作成する
GATK FastaAlternateReferanceMakerを使って、元となった参照配列に変異情報を当てはめる形で代替リファレンス配列(Fasta形式)を取得する。
感覚的にはVCF→FASTAに変換しているが、実際に変換したわけでは無く、VCFファイルに含まれる代替対立遺伝子(alternate allele)の配列をリファレンス配列と交換することで、変異を取り込んだ新しいFASTAを得ている。 ゲノムを新たにアセンブリしたわけではないので、利用には少し注意が必要。
自分で使用して失敗してしまった注意点もお伝えしますので、初めて使う方はぜひ読んでから開始することをお勧めします!
GATK Forumの説明を読んだだけで実行すると、十中八九同じ失敗をすると思います。
eupatho-bioinfomatics.hatenablog.com
SNPを使った解析の実践例 その1
eupatho-bioinfomatics.hatenablog.com
参考サイト
GATKの公式HP
https://gatk.broadinstitute.org/hc/en-us
初めてGATKに触れる時に、非常にお世話になったサイト
今後のGATK解析で使用するWGSデータの収集 -GATK解説シリーズ-part 2
sra-toolkit, fasterq-dump, sra-toolkit
導入難易度★☆☆☆☆
使用難易度★★☆☆☆
今回は何をする?
今後のGATK解析で使用するマラリア原虫のWGSデータを収集します。せっかくなので、以前のprefetchとは違うfasterq-dumpを使ったダウンロードの方法を紹介します。今後は、今回収集したデータを使って、GWASで使用する変異データの取得までを順番に紹介してきます。
私の使用感
以前の記事では、prefetch→fastq-dump→pigzを使って、sra形式→fastq形式→fastq.gz形式の順番でダウンロードしました。今回はfastq-dumpの後継ツールであるfasterq-dumpを使って、直接fastq形式でダウンロードします。また、fasterq-dumpではmulti-threadで処理できる点が優れています。ちなみに、以前まで使用していたWGSデータは、類似性が高すぎて使いづらかったため今後の解析には使用しない予定です。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog
1. 必要なツールをインストールする。
sra-toolkitおよびpigzを使用するので、必要であれば以前の記事を参照にインストールする。
eupatho-bioinfomatics.hatenablog.com
eupatho-bioinfomatics.hatenablog.com
2. Accession numberを羅列したテキストファイルを作成する。
2019年のScienceの論文で、アフリカのマラリア原虫の集団構造を2263株のWGSデータを使用して解析した大規模な研究のデータを一部拝借する。データの内訳は後日に必要なタイミングで説明するが、15カ国から2株ずつを選んだ。
まず、Accession numberを羅列したテキストファイルを用意する。
cat >SRA_list.txt <<EOF ERS032647 ERS032649 ERS347567 ERS347575 ERS199640 ERS199645 SRS399547 SRS378808 ERR015425 ERR015377 ERR063600 ERR211448 ERR484676 ERR343116 ERR562868 ERR580552 ERR450079 ERR450058 ERR405240 ERR405245 ERR1035536 ERR1045266 ERR636426 ERR636430 ERR701750 ERR701756 ERR1106528 ERR1106529 ERR2000569 EOF
4. 解析を実行する。
fasterq-dumpを実行する。fastq形式は非常に容量が大きくストレージを圧迫するため、pigzを使ってすぐにgz圧縮する。 なお、fasterq-dumpではpair-endとsingle-endを自動で認識するため、fastq-dumpで使用した--split-fileは使用しなくてよい。
while read line; do
fasterq-dump $line --outdir ./fastq --threads 16 --progress
pigz --processes 16 SRA/${line}_1.fastq
pigz --processes 16 SRA/${line}_2.fastq
done < SRA_list.txt
オプションについて
--outdir 出力ディレクトリ
--threads スレッド数 (デフォルト=6)
--progress 進捗状況の表示
--processes スレッド数 (デフォルト=8)
処理が正しく完了すると、
ERRXXXXXX_1.fastq.gz
ERRXXXXXX_2.fastq.gz
というファイルが得られる。
今回はこれで終わりです。
よければ他の記事のも見ていってください。
なお、本記事の執筆にあたりSRA Toolkitのfasta-dumpを高速化した fasterq-dumpを参考させていただきました。

GATKの導入-GATK解説シリーズ-part 1
GATKの導入とPATHの通し方
今回は何をする?
- GATK4を自分のPCに導入します。
GATKとは
GATKはBroad研究所が提供する、変異の検出に特化したゲノム解析ツールキットです。非常に多機能でゲノム解析分野で業界標準として広く使われていますが、操作難易度が高く、初心者は必ず躓くポイントになっています。GATKはもともとヒトゲノム解析用に開発されたものですが、現在ではあらゆる生物のゲノムデータを扱えるように進化しています。その範囲は、体細胞・生殖細胞のショートバリアント/インデル検出、コピー数(CNV)や構造変異(SV)への解析に及びます。また、GATKには、WGSの処理や品質管理などの関連作業を行うための多くのユーティリティが含まれており、頻繁に使われるPicardも搭載されています。また、GATKの読み方は(ジー・エー・ティー・ケイ)です。公式HPはこちら
また、本記事の内容はGetting started with GATK4の1-6に該当します。
GATK Best Practiceとは (GATK公式HPより引用・翻訳)
研究室でDNAを分離するとき、その作業を孤立したバラバラの作業のようには扱いません。すべての作業は、規定されたプロトコルのステップであり、収量と純度を最適化し、再現性とすべてのサンプルと実験における一貫性を確保するために慎重に取り扱われます。GATKは、シーケンシングデータの処理も同じように徹底的に行われるべきだという開発者らの理念に基づき、一連の作業を網羅したベストプラクティスのワークフローの使用が推奨されています。これは、ブロード研究所でテストされ、最も正確な結果を最も高い計算効率で得られるように最適化されています。
GATKを始めるには
GATKはLinuxまたはMacOSXで動作し、Windowsはサポートされていません。 Java 8 / JDK 1.8がインストールされていることを確認してください(OracleまたはOpenJDK、どちらでも構いません)。 GATKのパッケージはこちらから最新版(今回はGATK4.2.0.0)をダウンロードしてください。
ダウンロードしたファイルにインストールは必要ありません。ダウンロードしたパッケージを開き、jarファイルと起動スクリプトの入ったフォルダを、ハードディスクの作業ディレクトリに置くだけ使用することができます。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog
Local blastの使い方 [blastn]
コマンドラインでのblastの使い方
導入難易度★☆☆☆☆
使用難易度★★☆☆☆
このツールで何ができる?
BLAST(Basic Local Alignment Search Tool) は、類似性のある配列を持つ領域を見つけるツールです。このプログラムは、核酸やタンパク質の配列を任意の配列データベースと比較し、一致した部分の統計的有意性を計算します。BLASTは、配列間の機能的および進化的な関係を推測したり、ファミリー遺伝子の仲間を特定したりすることにも使えます。
私の使用感
BLAST にはNCBI Blastで利用できる ウェブ上のプラグラムがあります。ローカルで実施するBLASTでは、独自のデータベースを対象に設定することのできる点、大量のクエリ(解析する配列)を同時に処理することができる点、結果をその後の解析に組み込みやすい点など色々な長所があります。普段、バイオインフォマティクスを使っていない研究者の方でも、やってみると意外と簡単に実行できるので、ぜひ習得して役立てて下さい。
その他の詳細はMetagenomicsのHPを確認してみてください。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog
1. blastをインストールする。
conda install -c bioconda blast
インストールが正しく導入されたか確認する。
blastn --help
正しく導入されていれば、下記が表示される。
USAGE
blastn [-h] [-help] [-import_search_strategy filename]
[-export_search_strategy filename] [-task task_name] [-db database_name]
[-dbsize num_letters] [-gilist filename] [-seqidlist filename]
[-negative_gilist filename] [-entrez_query entrez_query]
[-db_soft_mask filtering_algorithm] [-db_hard_mask filtering_algorithm]
[-subject subject_input_file] [-subject_loc range] [-query input_file]
[-out output_file] [-evalue evalue] [-word_size int_value]
[-gapopen open_penalty] [-gapextend extend_penalty]
[-perc_identity float_value] [-qcov_hsp_perc float_value]
[-max_hsps int_value] [-xdrop_ungap float_value] [-xdrop_gap float_value]
[-xdrop_gap_final float_value] [-searchsp int_value]
[-sum_stats bool_value] [-penalty penalty] [-reward reward] [-no_greedy]
[-min_raw_gapped_score int_value] [-template_type type]
[-template_length int_value] [-dust DUST_options]
[-filtering_db filtering_database]
[-window_masker_taxid window_masker_taxid]
[-window_masker_db window_masker_db] [-soft_masking soft_masking]
[-ungapped] [-culling_limit int_value] [-best_hit_overhang float_value]
[-best_hit_score_edge float_value] [-window_size int_value]
[-off_diagonal_range int_value] [-use_index boolean] [-index_name string]
[-lcase_masking] [-query_loc range] [-strand strand] [-parse_deflines]
[-outfmt format] [-show_gis] [-num_descriptions int_value]
[-num_alignments int_value] [-line_length line_length] [-html]
[-max_target_seqs num_sequences] [-num_threads int_value] [-remote]
[-version]
DESCRIPTION
Nucleotide-Nucleotide BLAST 2.6.0+
・
・
・
* Incompatible with: gilist, seqidlist, negative_gilist, subject_loc,
num_threads
eupatho-bioinfomatics.hatenablog.com
続きを読む