Local blastの使い方 [blastn]
コマンドラインでのblastの使い方
導入難易度★☆☆☆☆
使用難易度★★☆☆☆
このツールで何ができる?
BLAST(Basic Local Alignment Search Tool) は、類似性のある配列を持つ領域を見つけるツールです。このプログラムは、核酸やタンパク質の配列を任意の配列データベースと比較し、一致した部分の統計的有意性を計算します。BLASTは、配列間の機能的および進化的な関係を推測したり、ファミリー遺伝子の仲間を特定したりすることにも使えます。
私の使用感
BLAST にはNCBI Blastで利用できる ウェブ上のプラグラムがあります。ローカルで実施するBLASTでは、独自のデータベースを対象に設定することのできる点、大量のクエリ(解析する配列)を同時に処理することができる点、結果をその後の解析に組み込みやすい点など色々な長所があります。普段、バイオインフォマティクスを使っていない研究者の方でも、やってみると意外と簡単に実行できるので、ぜひ習得して役立てて下さい。
その他の詳細はMetagenomicsのHPを確認してみてください。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog
1. blastをインストールする。
conda install -c bioconda blast
インストールが正しく導入されたか確認する。
blastn --help
正しく導入されていれば、下記が表示される。
USAGE
blastn [-h] [-help] [-import_search_strategy filename]
[-export_search_strategy filename] [-task task_name] [-db database_name]
[-dbsize num_letters] [-gilist filename] [-seqidlist filename]
[-negative_gilist filename] [-entrez_query entrez_query]
[-db_soft_mask filtering_algorithm] [-db_hard_mask filtering_algorithm]
[-subject subject_input_file] [-subject_loc range] [-query input_file]
[-out output_file] [-evalue evalue] [-word_size int_value]
[-gapopen open_penalty] [-gapextend extend_penalty]
[-perc_identity float_value] [-qcov_hsp_perc float_value]
[-max_hsps int_value] [-xdrop_ungap float_value] [-xdrop_gap float_value]
[-xdrop_gap_final float_value] [-searchsp int_value]
[-sum_stats bool_value] [-penalty penalty] [-reward reward] [-no_greedy]
[-min_raw_gapped_score int_value] [-template_type type]
[-template_length int_value] [-dust DUST_options]
[-filtering_db filtering_database]
[-window_masker_taxid window_masker_taxid]
[-window_masker_db window_masker_db] [-soft_masking soft_masking]
[-ungapped] [-culling_limit int_value] [-best_hit_overhang float_value]
[-best_hit_score_edge float_value] [-window_size int_value]
[-off_diagonal_range int_value] [-use_index boolean] [-index_name string]
[-lcase_masking] [-query_loc range] [-strand strand] [-parse_deflines]
[-outfmt format] [-show_gis] [-num_descriptions int_value]
[-num_alignments int_value] [-line_length line_length] [-html]
[-max_target_seqs num_sequences] [-num_threads int_value] [-remote]
[-version]
DESCRIPTION
Nucleotide-Nucleotide BLAST 2.6.0+
・
・
・
* Incompatible with: gilist, seqidlist, negative_gilist, subject_loc,
num_threads
eupatho-bioinfomatics.hatenablog.com
2. 解析の準備に必要な手順
本ブログの過去記事↓
eupatho-bioinfomatics.hatenablog.com
で取得したマラリア原虫のリファレンス配列(3D7_genomes/PlasmoDB-52_Pfalciparum3D7_Genome.fasta)を例として用いる。
まず、クエリ配列を記載したfastaファイルを準備する。
今回は、以前にも使用したPlasmodium falciparumのMAF1遺伝子の配列を用いる。
eupatho-bioinfomatics.hatenablog.com
上の記事の2を参照に、Genomic Sequence (UTRs highlighted)の塩基配列(ATG-TAA)をコピーする。
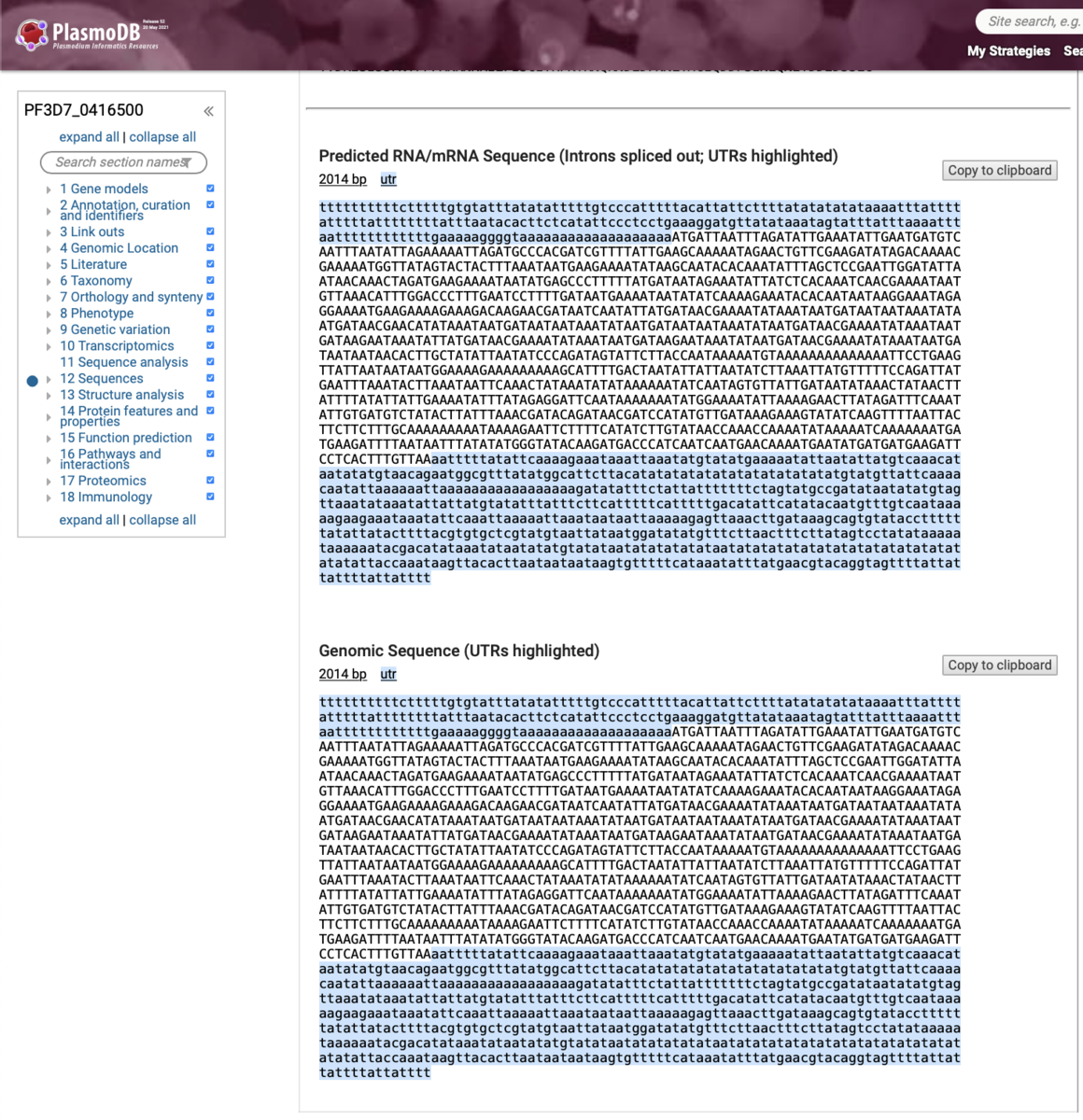
好きな方法で構わないが、120bpおきの配列を取得して、Fasta形式のテキストファイルとする。
例えば、指定した文字数でテキスト改行ツールであるCuttpicにコピーした配列を貼り付ける。
改行が間に挟まっている場合、位置がずれてしまうので改行は消去しておく。下にあるバーを動かして、任意の文字数を指定する。クリップマークを選択してコピーする。
最終的に、以下のfastaファイルをquery.fastaとして作業ディレクトリに保存する。
>MAF120 ATGATTAATTTAGATATTGAAATATTGAATGATGTCAATTTAATATTAGAAAAATTAGATGCCCACGATCGTTTTATTGAAGCAAAAATAGAACTGTTCGAAGATATAGACAAAACGAAAA >MAF240 ATGGTTATAGTACTACTTTAAATAATGAAGAAAATATAAGCAATACACAAATATTTAGCTCCGAATTGGATATTAATAACAAACTAGATGAAGAAAATAATATGAGCCCTTTTTATGATAA >MAF360 TAGAAATATTATCTCACAAATCAACGAAAATAATGTTAAACATTTGGACCCTTTGAATCCTTTTGATAATGAAAATAATATATCAAAAGAAATACACAATAATAAGGAAATAGAGGAAAAT >MAF480 GAAGAAAAGAAAGACAAGAACGATAATCAATATTATGATAACGAAAATATAAATAATGATAATAATAAATATAATGATAACGAACATATAAATAATGATAATAATAAATATAATGATAATA >MAF600 ATAAATATAATGATAACGAAAATATAAATAATGATAAGAATAAATATTATGATAACGAAAATATAAATAATGATAAGAATAAATATAATGATAACGAAAATATAAATAATGATAATAATAA >MAF720 CACTTGCTATATTAATATCCCAGATAGTATTCTTACCAATAAAAATGTAAAAAAAAAAAAAATTCCTGAAGTTATTAATAATAATGGAAAAGAAAAAAAAAGCATTTTGACTAATATTATT >MAF840 AATATCTTAAATTATGTTTTTCCAGATTATGAATTTAAATACTTAAATAATTCAAACTATAAATATATAAAAAATATCAATAGTGTTATTGATAATATAAACTATAACTTATTTTATATTA >MAF960 TTGAAAATATTTATAGAGGATTCAATAAAAAAATATGGAAAATATTAAAAGAACTTATAGATTTCAAATATTGTGATGTCTATACTTATTTAAACGATACAGATAACGATCCATATGTTGA >MAF1080 TAAAGAAAGTATATCAAGTTTTAATTACTTCTTCTTTGCAAAAAAAAATAAAAGAATTCTTTTCATATCTTGTATAACCAAACCAAAATATAAAAATCAAAAAAATGATGAAGATTTTAAT
3. データベースを作成する。
makeblastdbを使って、データベースを作成する。
#ゲノム配列は3D7_genomesの中に格納している。 makeblastdb -in 3D7_genomes/PlasmoDB-52_Pfalciparum3D7_Genome.fasta -dbtype nucl -parse_seqids
すると、以下のファイルが生成される。
ls 3D7_genomes PlasmoDB-52_Pfalciparum3D7_Genome.fasta.nhr PlasmoDB-52_Pfalciparum3D7_Genome.fasta.nsd PlasmoDB-52_Pfalciparum3D7_Genome.fasta.nin PlasmoDB-52_Pfalciparum3D7_Genome.fasta.nsi PlasmoDB-52_Pfalciparum3D7_Genome.fasta.nog PlasmoDB-52_Pfalciparum3D7_Genome.fasta.nsq
4. ローカルBlastを実行する。
今回は塩基配列を用いるので、blastnを使用する。なお、blastには以下のような機能がある。

blastn -db 3D7_genomes/PlasmoDB-52_Pfalciparum3D7_Genome.fasta -query query.fasta -out result.txt
-db 参照するデータベース名を指定する。
-query クエリ配列を入力したファイル名を指定する。
-out 結果を出力するファイル名を指定する。
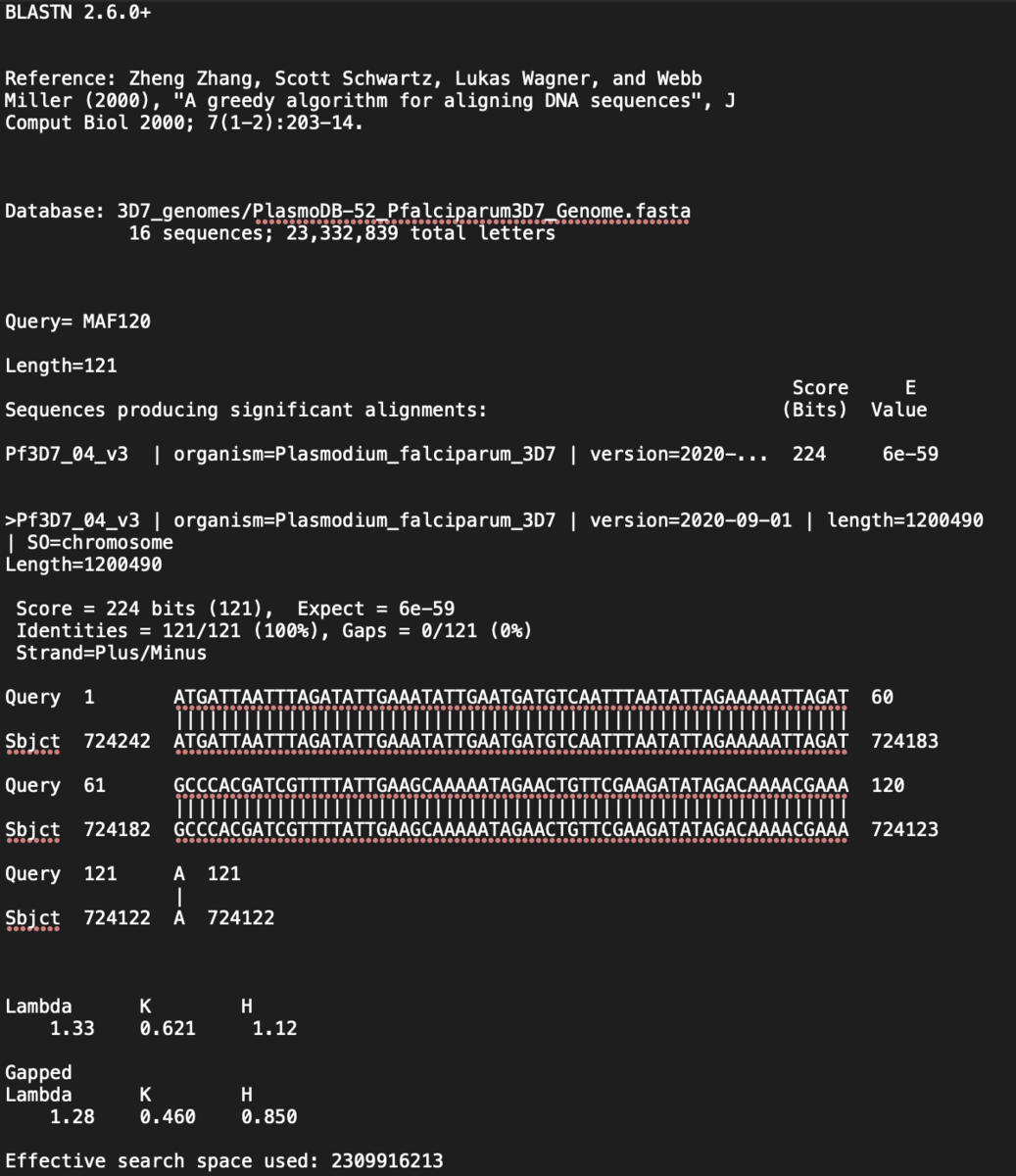
すると、result.txtが生成される。
5. outfmtで出力形式を変更する。
デフォルトの出力形式だと、後の解析に結果を使用しづらい。
そこでoutfmtを使用すると、さまざまな形式で結果を出力することができる。
*** Formatting options
-outfmt <String>
alignment view options:
0 = pairwise,
1 = query-anchored showing identities,
2 = query-anchored no identities,
3 = flat query-anchored, show identities,
4 = flat query-anchored, no identities,
5 = XML Blast output,
6 = tabular,
7 = tabular with comment lines,
8 = Text ASN.1,
9 = Binary ASN.1,
10 = Comma-separated values,
11 = BLAST archive format (ASN.1)
実際に-outfmt 6を実行してみると、
blastn -db 3D7_genomes/PlasmoDB-52_Pfalciparum3D7_Genome.fasta -query query.fasta -out result.txt -outfmt 6
MAF120 Pf3D7_04_v3 100.000 121 0 0 1 121 724242 724122 5.79e-59 224 MAF240 Pf3D7_04_v3 100.000 121 0 0 1 121 724121 724001 5.79e-59 224 MAF360 Pf3D7_04_v3 100.000 121 0 0 1 121 724000 723880 5.79e-59 224 MAF720 Pf3D7_04_v3 100.000 121 0 0 1 121 723637 723517 5.79e-59 224 MAF840 Pf3D7_04_v3 100.000 121 0 0 1 121 723516 723396 5.79e-59 224 MAF960 Pf3D7_04_v3 100.000 121 0 0 1 121 723395 723275 5.79e-59 224 MAF1080 Pf3D7_04_v3 100.000 121 0 0 1 121 723274 723154 5.79e-59 224
となる。この場合、クエリ配列名、一致した塩基数、データベース上での領域などが羅列したファイルが得られる。
お疲れ様でした、今回はこれで終わりです。
よければ他の記事も見ていってください。
