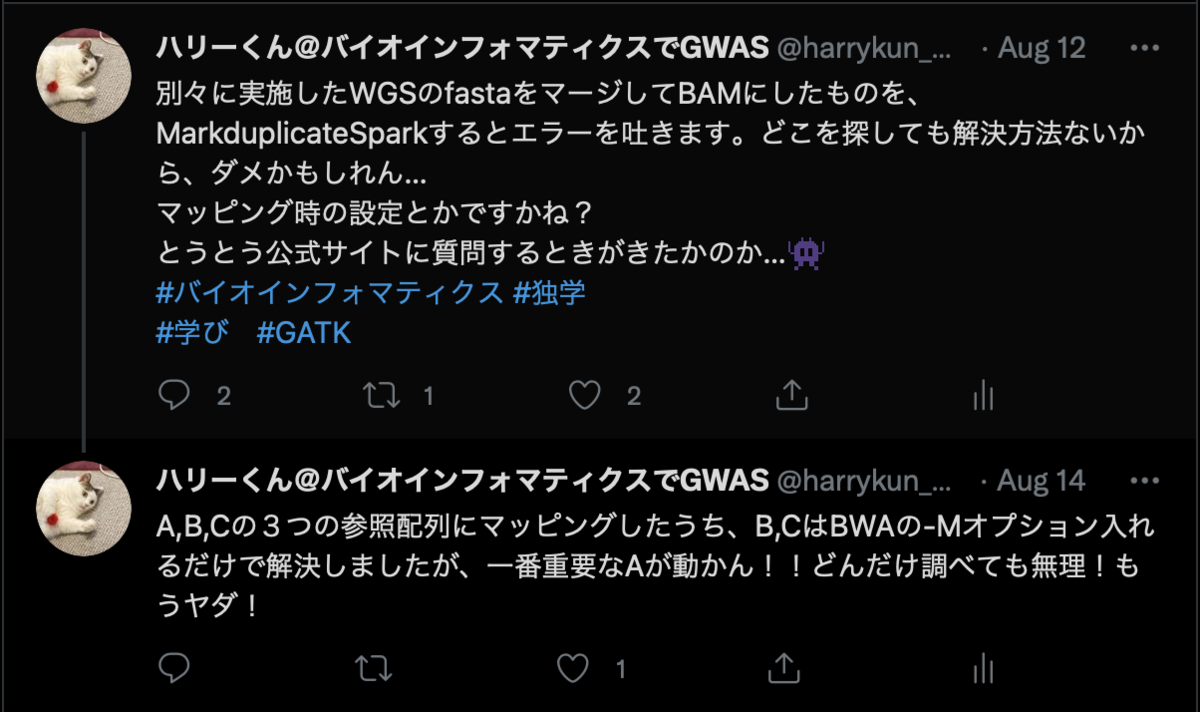
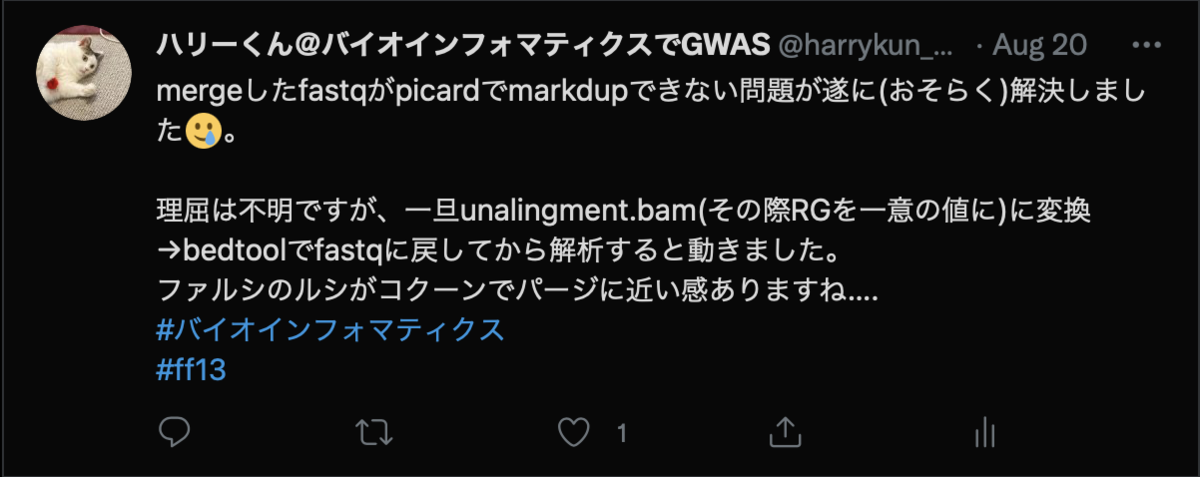
マージしたFastqを使ってMarkduplicateを実行するのに苦労した話
使用するツール Cat, Trimmomatic, Picard FastqToSam, Bedtools bamtofastq
今回は何をする?
最近、カバレッジの不足を補うために、別々に実施したWGSデータを合体させて解析に使う機会がありました。
「単純にcatコマンドで合成するだけでできるよ」と共同研究者が教えてくれたのですが、私の場合はうまくいかず解決するために最終的に2週間もかかってしまったので、
同じ悩みを抱えた方に向けて記録を残しておきます。
↓ツイッターログ
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog
本ブログのメインコンテンツのまとめ記事は↓こちら
eupatho-bioinfomatics.hatenablog.com
続きを読むGATK FastaAlternateReferanceMakerを使って代替参照配列を作成する
GATK FastaAlternateReferanceMakerの使い方
使用難易度★★★★☆
本記事は、GATK解説シリーズのPart 11です。
GATK解説シリーズのリンクまとめは↓こちら
GATKの導入方法から、変異情報の取得までをハプロイドの病原体を使った実例とともに紹介しています。
eupatho-bioinfomatics.hatenablog.com
今回は何をする?
GATK FastaAlternateReferanceMakerを使って、元となった参照配列に変異情報を当てはめる形で代替リファレンス配列(Fasta形式)を取得する。
感覚的にはVCF→FASTAに変換しているが、実際に変換したわけでは無く、VCFファイルに含まれる代替対立遺伝子(alternate allele)の配列をリファレンス配列と交換することで、変異を取り込んだ新しいFASTAを得ている。 ゲノムを新たにアセンブリしたわけではないので、利用には少し注意が必要。
自分で使用して失敗してしまった注意点もお伝えしますので、初めて使う方はぜひ読んでから開始することをお勧めします!
GATK Forumの説明を読んだだけで実行すると、十中八九同じ失敗をすると思います。
fastaファイルをコマンドラインで操作するseqkitも少しだけ紹介する。
vcftoolsもしくはbcftoolsでも似たような機能が利用できる。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。 フォローする @harrykun_blog
GATK公式HPのFastaAlternateReferanceMakerのリンク
RAxML-ngによるSNP系統解析の実践方法 [最尤法] [maximum likelihood]
主な使用ツール; RAxML-NG, Modeltest-NG, FigTree
使用難易度★★★☆☆
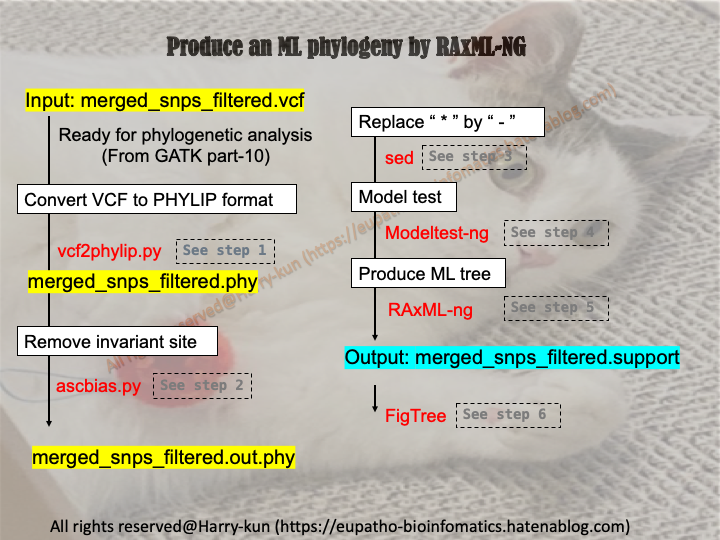
今回は何をする?
- 前回の記事で取得したSNP情報を基に最尤法による系統解析を行います。
全ゲノムスケールのSNP情報を使用することで、高精度な系統解析が実現可能です。
ここではWGSを使用していますが、もちろんもっと短いDNA配列やタンパク質にも応用できます。
2つのpythonベースのプログラム
vcf2pylip.py,ascbias.pyを使って、vcfファイルを系統解析用のPHYLIP形式に調整します。コマンドラインでの実装が簡単・迅速に実行できるRAxML-ngを使用して、Boostrap数を指定して系統解析を行います。
フリーソフトのFigTreeを使って系統樹を描写します。
GUIで操作できるMEGA Xを使った手法も別の記事で紹介しています。
ブログ主は普段、MEGA Xはアライメント結果の視覚的な確認用にして、系統解析にはRAxML-ngを使っています。
RAxMLの時は、コマンドが分かりづらくで使いづらかったのですが、NG(Next Generation)になって大幅に使いやすくなっています。
eupatho-bioinfomatics.hatenablog.com
本ブログでは、バイオインフォマティクスを使った解析方法を実践的に紹介しています。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog
- なお、この記事の方法は、Macの使用を前提にしています。
持っていない方は買いましょう(笑)。最低でも16GB以上のメモリがないとしんどいと思います。(有ればあるほどよい)
決して安くはないですが、、Mac ライフは最高ですよ♪
 - スペースグレイ")
GATK BQSR後のVariant Calling -GATK解説シリーズ-part 10
GATK BQSR後のVariant Calling
使用難易度★☆☆☆☆
本記事は、GATK解説シリーズのPart 10です。
GATK解説シリーズのリンクまとめは↓こちら eupatho-bioinfomatics.hatenablog.com
今回は何をする?
前回のGATK VariantFilterationで出力された、
*.bqsr.bamを基に2回目のVariant Callingを行います。今回行う内容は、GATK解説シリーズ Part 5 からPart 8で行った仕事の繰り返しになります。新しく覚えることは特にありません。
今回得た
merged_snps/indels_filtered.vcfを使って、多様なゲノムワイド解析を行うことができます。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog