RAxML-ngによるSNP系統解析の実践方法 [最尤法] [maximum likelihood]
主な使用ツール; RAxML-NG, Modeltest-NG, FigTree
使用難易度★★★☆☆
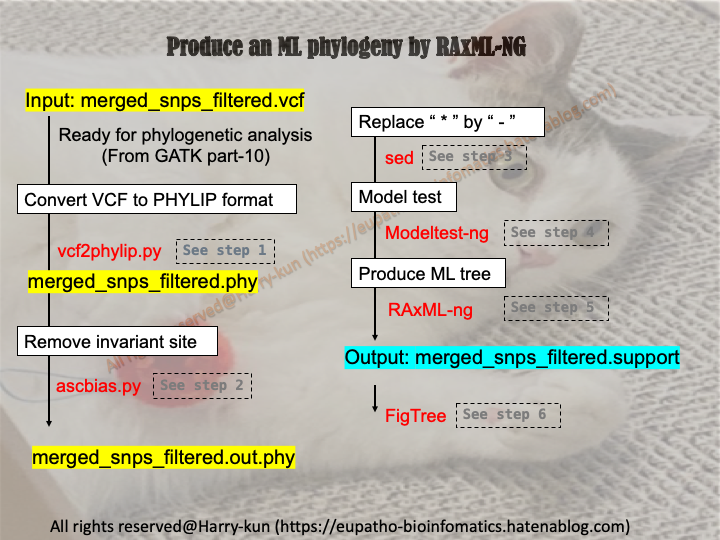
今回は何をする?
- 前回の記事で取得したSNP情報を基に最尤法による系統解析を行います。
全ゲノムスケールのSNP情報を使用することで、高精度な系統解析が実現可能です。
ここではWGSを使用していますが、もちろんもっと短いDNA配列やタンパク質にも応用できます。
2つのpythonベースのプログラム
vcf2pylip.py,ascbias.pyを使って、vcfファイルを系統解析用のPHYLIP形式に調整します。コマンドラインでの実装が簡単・迅速に実行できるRAxML-ngを使用して、Boostrap数を指定して系統解析を行います。
フリーソフトのFigTreeを使って系統樹を描写します。
GUIで操作できるMEGA Xを使った手法も別の記事で紹介しています。
ブログ主は普段、MEGA Xはアライメント結果の視覚的な確認用にして、系統解析にはRAxML-ngを使っています。
RAxMLの時は、コマンドが分かりづらくで使いづらかったのですが、NG(Next Generation)になって大幅に使いやすくなっています。
eupatho-bioinfomatics.hatenablog.com
本ブログでは、バイオインフォマティクスを使った解析方法を実践的に紹介しています。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog
- なお、この記事の方法は、Macの使用を前提にしています。
持っていない方は買いましょう(笑)。最低でも16GB以上のメモリがないとしんどいと思います。(有ればあるほどよい)
決して安くはないですが、、Mac ライフは最高ですよ♪
 - スペースグレイ")
今回使用するVCFファイルについて
解析に使用する自分のデータがない方は、本ブログのGATK解説シリーズで作成した25株のPlasmodium falciparum(マラリアを起こす病原体)の全ゲノムデータを使って検出したSNPファイルをダウンロードしてください。
こちらのリンクからダウンロードできます(200MBほど)
VCFファイルについて知りたい方は↓こちらの記事で分かりやすく説明しています。
eupatho-bioinfomatics.hatenablog.com
eupatho-bioinfomatics.hatenablog.com
生のWGS(fastp)から変異情報を検出してVCFファイルの作成までの工程は、GATK解説シリーズで詳しく解説しています。
GATK解説シリーズのリンクまとめは↓こちら
eupatho-bioinfomatics.hatenablog.com
Step 0 マルチアレルなバリアントを除く。(必要に応じて実施)
VCFファイルにMultiAllelic(1箇所のバリアントサイトに複数の代替対立遺伝子を持つ)なバリアントが含まれているとエラーがでるので、GATK SelectVariantsを使ってSNPを選別します。
SelectVariantsで必要なPlasmodium falciparum 3D7のリファレンスゲノムの入手方法は別の記事で紹介しています。
index=PlasmoDB-52_Pfalciparum3D7_Genome.fasta
gatk SelectVariants \
-R ${index} \
-V merged_snps_filtered.vcf \
-O merged_snps_filtered.vcf \
--restrict-alleles-to BIALLELIC
GATKをまだ導入していない方は、こちらの記事を参考にしてください。
eupatho-bioinfomatics.hatenablog.com
GATK SelectsVariantの使い方はこちらで詳しく解説しています。
eupatho-bioinfomatics.hatenablog.com
Step 1 VCFをPHYLIP形式に変換する。
vcf2pylip.pyを使って、VCF形式の変異情報をPHYLIPアライメントに変換します。
python vcf2phylip.py --input merged_snps_filtered.vcf
正しく終了すると、merged_snps_filtered.min4.phyが生成される。
一応、GATKの導入が上手くいかなかった方のために、このファイルもリンクを貼っておきます。
vcf2pylip.pyの使い方は、こちらの記事で詳しく紹介しています。
eupatho-bioinfomatics.hatenablog.com
Step 2 Invariant siteを取り除く
ascbias.pyを使って、invariant siteを取り除きます。
Githubのリンク(https://github.com/btmartin721/raxml_ascbias)からascbias.pyをダウンロードして、使用するディレクトリに保存する。
ascbiasを実行する。
python ascbias.py -p merged_snps_filtered.min4.phy --outfile merged_snps_filtered_out.phy
正しく終了すると、merged_snps_filtered_out.phyが生成される。
Step 3 PHYLIP中の*を-に変換する。
RAxMLを実施する前に、PHYLIPファイル中に*が含まれているとエラーが出る場合があるので、-に変換する。
テキストファイルをコマンドラインで編集するsedを使う。
cat merged_snps_filtered_out.phy | sed -e "s/*/-/g" > merged_snps_filtered_out_2.phy
Step 4 最適なモデルを推計する。
ModelTest-NGを使用して、最適な進化モデルを推定する。
1. ModelTest-NGをインストールする。
conda install -c bioconda modeltest-ng
2. ModelTest-NGを実行する。
modeltest-ng -i merged_snps_filtered_out_2.phy
以下のようなプログラムが走る。
_ _ _ _ _ _ _____
| | | | | | | | \ | |/ ____|
_ __ ___ ___ __| | ___| | |_ ___ ___| |_ | \| | | __
| '_ ` _ \ / _ \ / _` |/ _ \ | __/ _ \/ __| __| | . ` | | |_ |
| | | | | | (_) | (_| | __/ | || __/\__ \ |_ | |\ | |__| |
|_| |_| |_|\___/ \__,_|\___|_|\__\___||___/\__| |_| \_|\_____|
--------------------------------------------------------------------------------
modeltest x.y.z
Copyright (C) 2017 Diego Darriba, David Posada, Alexandros Stamatakis
License GPLv3+: GNU GPL version 3 or later <http://gnu.org/licenses/gpl.html>.
This is free software: you are free to change and redistribute it.
There is NO WARRANTY, to the extent permitted by law.
Written by Diego Darriba.
--------------------------------------------------------------------------------
Physical cores: 4
Logical cores: 8
Memory: 16GB
Extensions: AVX
・
・
・
完了すると、merged_snps_filtered_out2.phy.outが生成される。
長文のログが保存されるが、
Best model according to BIC Best model according to AIC Best model according to AICc
の欄に最適なモデルが記載される。
例えば、AICは以下のような感じ
Best model according to AIC --------------------------- Model: GTR+G4 lnL: -3539028.1044 Frequencies: 0.2952 0.2003 0.2030 0.3015 Subst. Rates: 1.0070 2.2096 1.4433 1.2127 2.2250 1.0000 Inv. sites prop: - Gamma shape: 2.3682 Score: 7078168.2088 Weight: 0.7076 --------------------------- Parameter importances --------------------------- P.Inv: - Gamma: 0.7076 Gamma-Inv: 0.2924 Frequencies: 1.0000 --------------------------- Model averaged estimates --------------------------- P.Inv: - Alpha: 2.3682 Alpha-P.Inv: 2.3737 P.Inv-Alpha: 0.0000 Frequencies: 0.2952 0.2003 0.2029 0.3015 Commands: > phyml -i merged_snps_filtered_out2.phy -m 012345 -f m -v 0 -a e -c 4 -o tlr > raxmlHPC-SSE3 -s merged_snps_filtered_out2.phy -m GTRGAMMAX -n EXEC_NAME -p PARSIMONY_SEED > raxml-ng --msa merged_snps_filtered_out2.phy --model GTR+G4 > paup -s merged_snps_filtered_out2.phy > iqtree -s merged_snps_filtered_out2.phy -m GTR+G4
簡単に調べてみましたが、BIC, AIC, AICcのどれが最適ななのかはよく分かりませんでした、AIC > BICのような記述はありましたが。。。
Step 5 最尤ツリーを計算する。
RAxML-ngを使って、最尤ツリーの描写に必要なファイルを得る。
1. RAxML-NGをインストールする。
conda install -c bioconda raxml-ng
2. RAxML-ng-NGを実行する。
注意:ものすごい時間かかりますので、テストデータを使って実行しないでください。
解析結果は下にリンクを用意しています
raxml-ng --msa merged_snps_filtered_out2.phy --model GTR+G4+ASC_LEWIS --all --bs-trees 1000 --threads 16
オプションについて
--msa; 解析を行うファイルのファイル名を指定する。
--all; 樹形探索とブートストラップ解析を行う。
--model; modeltestで求めたモデル名を入力する。
--bs-trees ブートストラップ数を指定する。論文でよく見るのは1000ですが、全ゲノム規模の系統解析で実行するとかなり長時間かかります。(240Gのメモリで10日間とか)
--threads 解析に使うスレッド数を指定する。必ず、最大スレッド数以下の数値を入力すること。
完了すると生成される、merged_snps_filtered_out2.supportを使って、系統樹を作成します。
解析の時間ががもったいないと思いますので、解析結果を貼っておきます。こちらからダウンロードしてください。
Step 6 ツリーを描写する。
FigTreeを使ってMLツリーを取得できます。FigTreeはGUIのフリーソフトです。
- こちらのリンクから
FigTree.v1.4.4.dmgをダウンロードする。
- FigTreeを起動する。
File→Openを選び、解析後に出力されたmerged_snps_filtered_out2.supportを開く。
開く際に、解析に使用した変数名を指定するように言われるので適当にいれておく。(今回はBSとした)
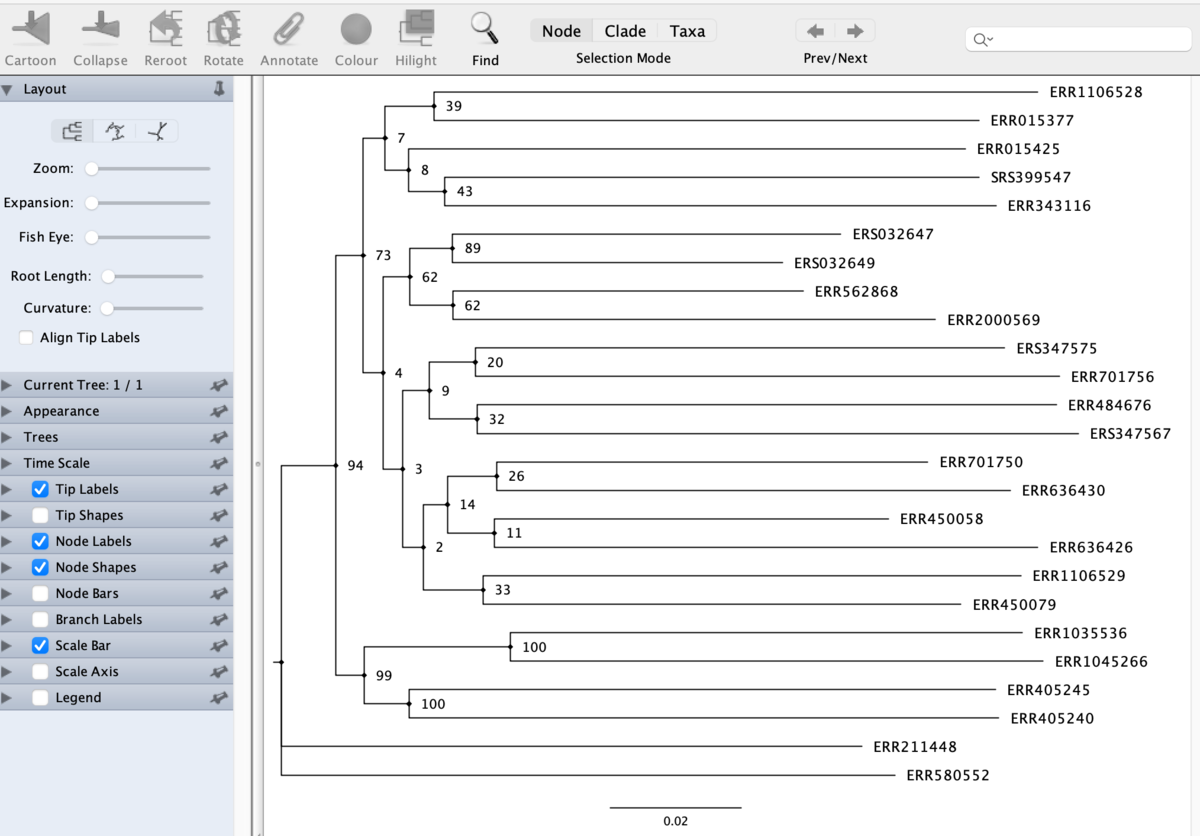
FigTreeの詳細な使い方は、他を参照してもらいたいが、今回はBoostrap値を表示する。
左側のNode Labelsを開き、BSを選択すると、枝の分かれ目にBoostrap値が表示される。
画像の出力は、File→Export 好きな形式を選択する。
解析は以上で終わりです。
最後に、今回の系統図を少し解説しておきましょう。
各サンプルは以下のように、アフリカ大陸のマラリア患者から採取された原虫です。
原著では数千株のデータを用いた大規模なPCAなどが見れますが、そこで地理的および媒介者などの要因でPlasmodiumが遺伝的に分断されたクラスターを形成していることが示されています。
サンプルの由来
Geography Accession=Number Malawi ERS032647 Malawi ERS032649 DRCongo ERS347567 DRCongo ERS347575 Senegal SRS399547 Mali ERR015425 Mali ERR015377 Guinea ERR211448 Ghana ERR484676 Ghana ERR343116 Cameroon ERR562868 Cameroon ERR580552 Ghana ERR450079 Ghana ERR450058 Tanzania ERR405240 Tanzania ERR405245 Ethiopia ERR1035536 Ethiopia ERR1045266 Cote_DIvoire ERR636426 Cote_DIvoire ERR636430 Kenya ERR701750 Kenya ERR701756 Gambia ERR1106528 Gambia ERR1106529 Gabon ERR2000569
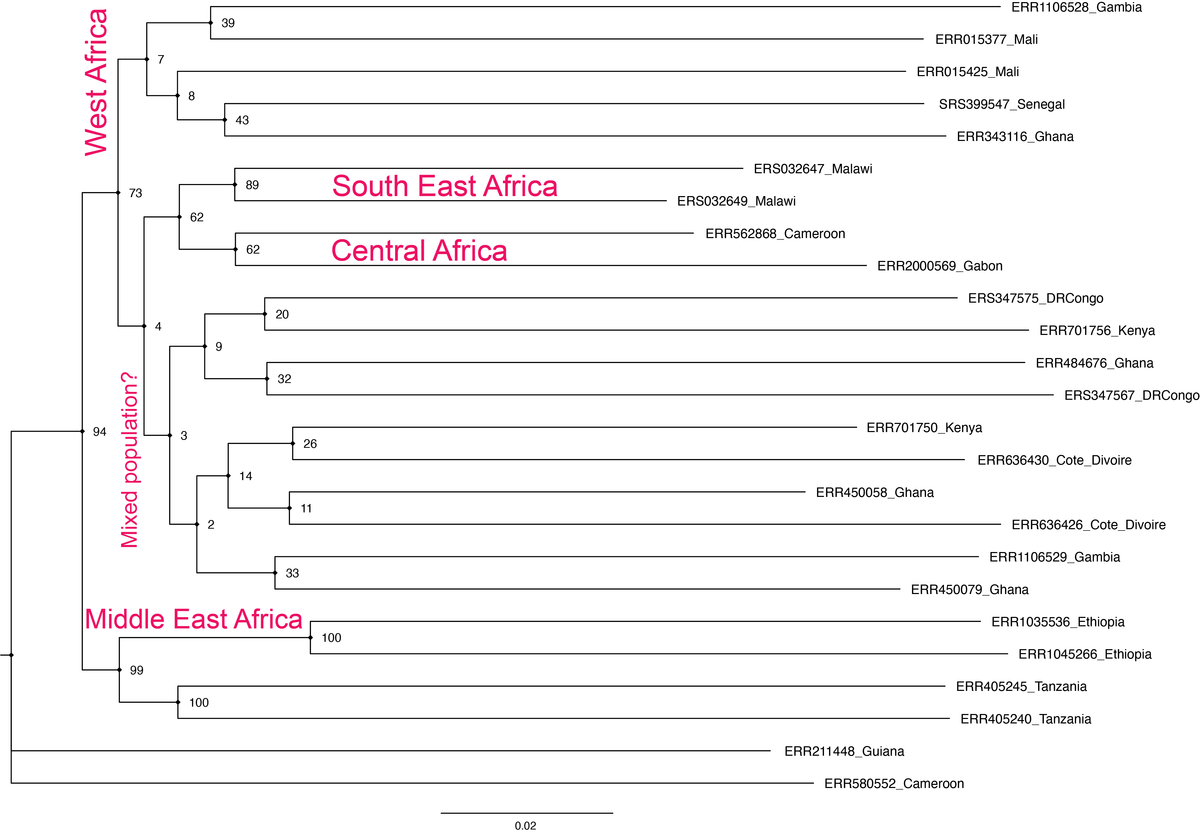
今回の結果でも、おなじように西、中央、東アフリカでまとまっていることから、似たような結果を得られていることが分かります。
(サンプル数が本来の1%とごく少数なためか、低BSの混合した集団もいますが・・・実は別の理由もあるんですがそれは置いておきましょう)
PCA解析も、需要がありそうであれば再現してみようと思います。
お疲れ様でした。今回はこれで終わりです。
少しでも解析の手助けになれれば幸いです。
なお、本記事の作成には【SNP 解析】ML系統樹作成 https://nemunemu-nyanko.hatenablog.com/entry/2020/12/28/173117
を参考にさせていただきました。