マージしたFastqを使ってMarkduplicateを実行するのに苦労した話
使用するツール Cat, Trimmomatic, Picard FastqToSam, Bedtools bamtofastq
今回は何をする?
最近、カバレッジの不足を補うために、別々に実施したWGSデータを合体させて解析に使う機会がありました。
「単純にcatコマンドで合成するだけでできるよ」と共同研究者が教えてくれたのですが、私の場合はうまくいかず解決するために最終的に2週間もかかってしまったので、
同じ悩みを抱えた方に向けて記録を残しておきます。
↓ツイッターログ
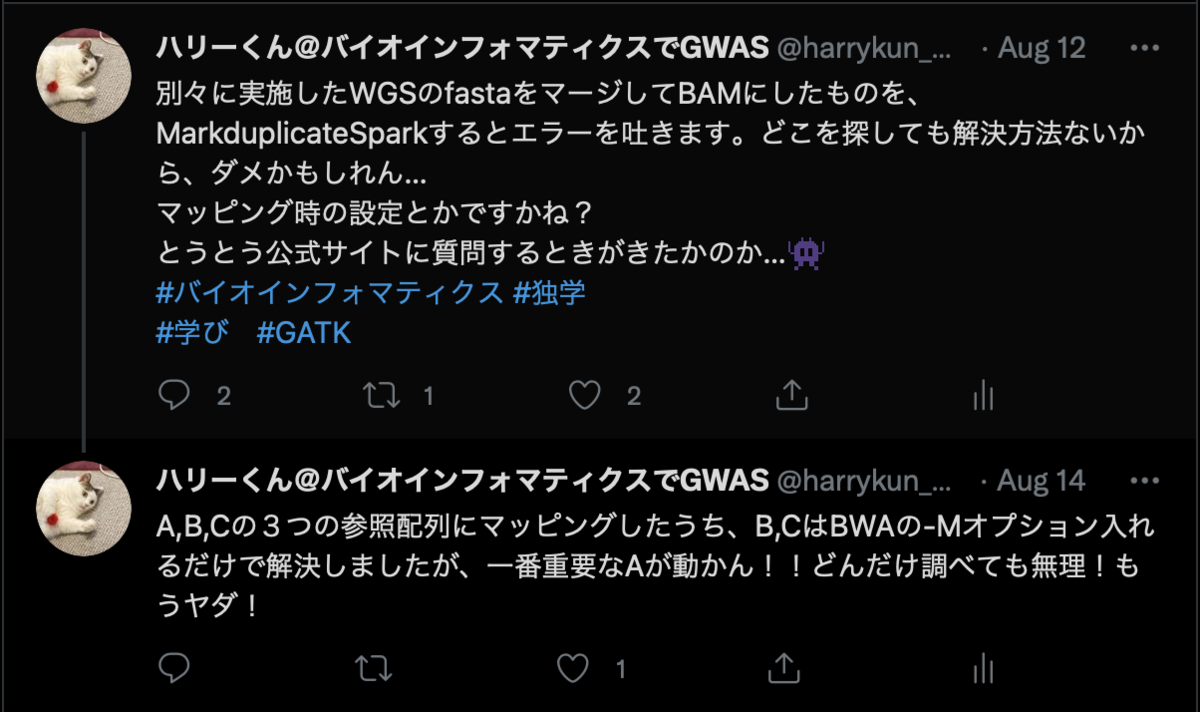
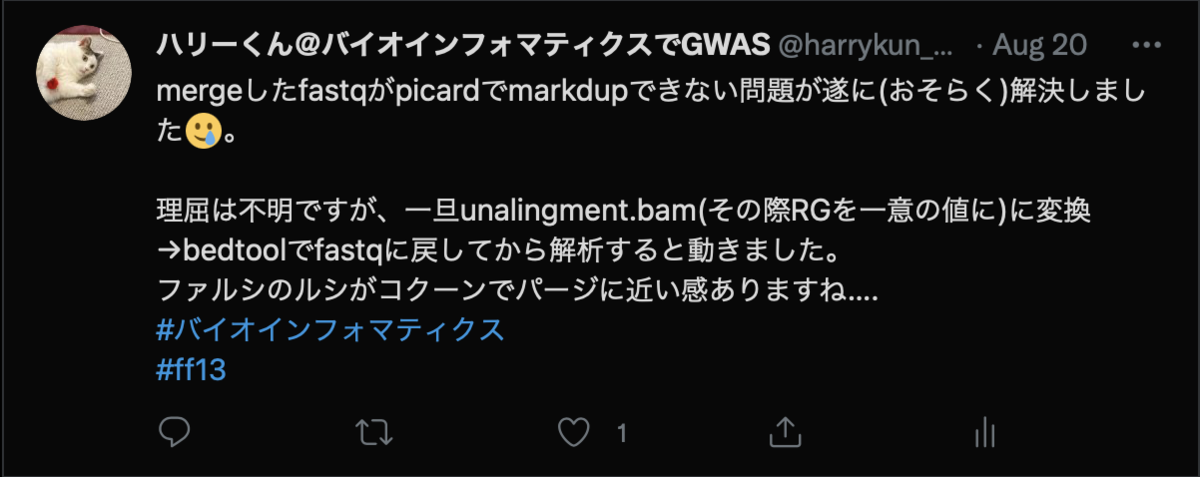
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog
本ブログのメインコンテンツのまとめ記事は↓こちら
eupatho-bioinfomatics.hatenablog.com
ステップ1 Catコマンドで複数のFastqファイルを連結する。
サンプルAについて、2回分のペアエンド (R1とR2) Fastqファイルが手元にあると仮定します。
Sample A1; A1_R1.fastq.gz A1_R2.fastq.gz
Sample A2; A2_R1.fastq.gz A2_R2.fastq.gz
これらのファイルをエンドごとにマージした新しいFastqファイルを作成します。
A_Merged_R1.fastq.gz,とA_Merged_R2.fastq.gz
cat A1_R1.fastq.gz A2_R1.fastq.gz > A_Merged_R1.fastq.gz cat A1_R2.fastq.gz A2_R2.fastq.gz > A_Merged_R2.fastq.gz
ステップ2 Trimmmomaticでトリミングする。
A_Merged_R1.fastq.gzとA_Merged_R2.fastq.gzからトリミング後に、 A_Merged_cleaned_1.fastq.gzとA_Merged_cleaned_2.fastq.gzを得ます。
Trimommaticについては過去記事を参照してください。
eupatho-bioinfomatics.hatenablog.com
ステップ3 Picard FastqToSamで、マッピングされていないBAM形式に変換する。
この操作で、Fastqのリード属性を統一する。 FastqToSamについてはこちらのリンク(外部HP)を参照。
この工程で、A_Merged_unaligned_reads.bamが得られる。
java -jar picard.jar FastqToSam \
F1=A_Merged_cleaned_1.fastq.gz \
F2=A_Merged_cleaned_2.fastq.gz \
O=A_Merged_unaligned_reads.bam \
SM=A \
RG=A
ステップ4 Bedtools bamtofastqで、再度Fastq形式に変換する。
bedtools bamtofastq -i A_unaligned_reads.bam -fq A_1.fastq -fq2 A_2.fastq
ステップ5 BWA memで -M オプションを指定してマッピングする。
BWA memを使ってMappingする。
理屈はよく分かりませんが、ここで-mオプションを指定しておきます。
bamRG="@RG\tID:L\tSM:"A"\tPL:illumina\tLB:lib1\tPU:unit1"
bwa mem -M -t 16 -R ${bamRG} Reference.fasta \
A_1.fastq A_2.fastq > A.sam
ステップ6 Markduplicate
最後に、Picard Markduplicateを実行します。
これも原因は不明ですが、Sparkバージョンだとエラーがでるのでノーマルバージョンで実行します。
samtools sort -@ 16 (スレッド数) A.sam > A.bam
gatk MarkDuplicates \
-I A.bam \
-O A.markdup.bam \
-M A.markdup.metrics.txt \
--CREATE_INDEX
最後に、A.markdup.bamが出力されます。
このファイルを使って、以後のVariant Callを実行します。
マージ前後のリード数、カバレッジなどからうまく動作していることは確認したので、
多分大丈夫だと思います。
お疲れ様でした。今回はこれで終わりです。
よければ他の記事のも見ていってください。
系統解析について解説した記事は↓こちら
eupatho-bioinfomatics.hatenablog.com
eupatho-bioinfomatics.hatenablog.com
VCFファイルについて解説した記事は↓こちら
eupatho-bioinfomatics.hatenablog.com
バイオインフォマティクス関連の書籍紹介は↓こちら