VCFファイルとはなにかを説明します-前編
VCF (Variant call format)ファイルの見方
今回は何をする?
- これまでに全くVCFファイルに触れたことのない方に向けて、ファイルを構成する要素を詳しく解説します。
- VCFは変異解析に不可欠な要素であり、必ず理解しなければなりません。 初めて見た時の感想は、「うわ、ちんぷんかんぶん・・・」という印象を持つと思いますが、慣れてくると意外と単純な要素で構成されていることに気が付きます。VCFファイルの形式を理解しないと、GATKやvcf/bcftoolsを使ったフィルタリングで何をしているか理解できなくなってしまうので、この機会に一緒に勉強していきましょう。
- 二つの記事を読み終わる頃には、きっとVCFファイルに対する苦手意識は無くなっていると思います!前編では、VCFを構成する要素について順番に解説します。後編では各バリアントサイトのレコードに付随する項目やスコアについて説明します。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。 フォローする @harrykun_blog
今回はこちらで用意したVCFファイルをもとに説明していきます。すべてを貼り付けると長大になってしまうため、一部を抜粋します。 全体を見たい方はこちらのリンクからダウンロードしてください。(400MBほど)
GATK公式HPのVCFについてのリンク
1. VCFフォーマットとは
VCF(Variant Call Format)を一言で説明すると、SNP、インデル、構造的変異を記述するために標準化されたテキストファイル形式です。
フォーマットの詳細な仕様は、こちらのPDFで見ることができます。
VCFはGATKがバリアントコールに使用しているフォーマットで、主にバリアントの種類とシークエンス、および個々のバリアントに関する複数サンプルのジェノタイプについての情報が記述されます。とはいえ、GATK HaplotypeCallerのようなツールが生成するVCFファイルは少し複雑です。ここでは、GATKが出力するVCFファイルを理解するために知っておくべき点について説明します。
なお、VCFファイルはプレーンテキストファイルなので、Excelなどのテキストエディタで開いて編集することができますが、VCFファイルは大きな容量になりがちであるためファイルを読み込むのに時間がかかる場合があります。そのため、GATKのSelectVariantsのようなツールを使ってデータを編集するアプローチをとる必要がでてくるわけです。
また、Microsoft Wordなどのワープロでvcfを編集するとフォーマットが崩れてしまうので絶対にしないでください。
2. VCFファイルの構成
VCFファイルは、ヘッダーとバリアントコールレコードの2つの要素で構成されています。
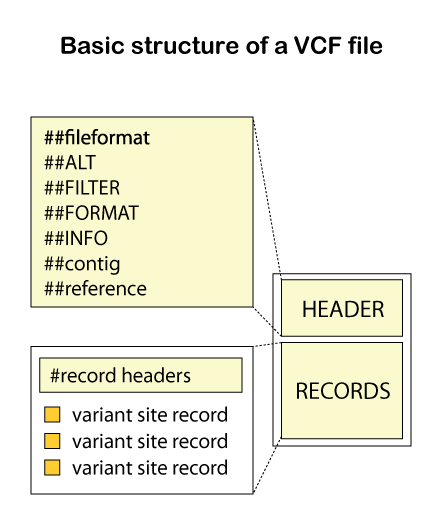
実際のデータを順番に見ていきましょう。
VCFバージョンの情報
1行目は、ファイルが準拠しているVCFのバージョンを示します。バイオインフォマティクスは変化の激しい分野であり、ファイルのフォーマットも急速に進化しています。VCFファイルを解析しようとして予期せぬ問題に遭遇した場合、フォーマットの仕様変更がないか、バージョンを確認してください。
##fileformat=VCFv4.2
FILTERについて
FILTER行では、データにどのようなフィルターが適用されたかが記述されています。下の例では、FILTER=<ID=LowQual,Description="Low quality">というフィルターが適用されています。 ここに記載されたフィルターに適合しなかったレコードのFILTERフィールドには、フィルタのID(ここではLowQual)が記載されます。 羅列されたすべてのフィルター条件をすべて満たす場合にPASSと記載されます。
##FILTER=<ID=LowQual,Description="Low quality"> ##FILTER=<ID=PASS,Description="All filters passed">
FORMAT および INFO について
これらの行は、VCFファイルのFORMATおよびINFO列に含まれるアノテーションが記述されています。
簡単に言うと、FORMATが個別サンプルに付随する情報で、INFOが全体に付随する情報になります。
各項目の詳細は、後半に記載します。
FORMAT
##FORMAT=<ID=AD,Number=R,Type=Integer,Description="Allelic depths for the ref and alt alleles in the order listed"> ##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Approximate read depth (reads with MQ=255 or with bad mates are filtered)"> ##FORMAT=<ID=GQ,Number=1,Type=Integer,Description="Genotype Quality"> ##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype"> ##FORMAT=<ID=MIN_DP,Number=1,Type=Integer,Description="Minimum DP observed within the GVCF block"> ##FORMAT=<ID=PL,Number=G,Type=Integer,Description="Normalized, Phred-scaled likelihoods for genotypes as defined in the VCF specification"> ##FORMAT=<ID=RGQ,Number=1,Type=Integer,Description="Unconditional reference genotype confidence, encoded as a phred quality -10*log10 p(genotype call is wrong)"> ##FORMAT=<ID=SB,Number=4,Type=Integer,Description="Per-sample component statistics which comprise the Fisher's Exact Test to detect strand bias.">
INFO
##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele count in genotypes, for each ALT allele, in the same order as listed"> ##INFO=<ID=AF,Number=A,Type=Float,Description="Allele Frequency, for each ALT allele, in the same order as listed"> ##INFO=<ID=AN,Number=1,Type=Integer,Description="Total number of alleles in called genotypes"> ##INFO=<ID=BaseQRankSum,Number=1,Type=Float,Description="Z-score from Wilcoxon rank sum test of Alt Vs. Ref base qualities"> ##INFO=<ID=DP,Number=1,Type=Integer,Description="Approximate read depth; some reads may have been filtered"> ##INFO=<ID=END,Number=1,Type=Integer,Description="Stop position of the interval"> ##INFO=<ID=ExcessHet,Number=1,Type=Float,Description="Phred-scaled p-value for exact test of excess heterozygosity"> ##INFO=<ID=FS,Number=1,Type=Float,Description="Phred-scaled p-value using Fisher's exact test to detect strand bias"> ##INFO=<ID=InbreedingCoeff,Number=1,Type=Float,Description="Inbreeding coefficient as estimated from the genotype likelihoods per-sample when compared against the Hardy-Weinberg expectation"> ##INFO=<ID=MLEAC,Number=A,Type=Integer,Description="Maximum likelihood expectation (MLE) for the allele counts (not necessarily the same as the AC), for each ALT allele, in the same order as listed"> ##INFO=<ID=MLEAF,Number=A,Type=Float,Description="Maximum likelihood expectation (MLE) for the allele frequency (not necessarily the same as the AF), for each ALT allele, in the same order as listed"> ##INFO=<ID=MQ,Number=1,Type=Float,Description="RMS Mapping Quality"> ##INFO=<ID=MQRankSum,Number=1,Type=Float,Description="Z-score From Wilcoxon rank sum test of Alt vs. Ref read mapping qualities"> ##INFO=<ID=QD,Number=1,Type=Float,Description="Variant Confidence/Quality by Depth"> ##INFO=<ID=RAW_MQandDP,Number=2,Type=Integer,Description="Raw data (sum of squared MQ and total depth) for improved RMS Mapping Quality calculation. Incompatible with deprecated RAW_MQ formulation."> ##INFO=<ID=ReadPosRankSum,Number=1,Type=Float,Description="Z-score from Wilcoxon rank sum test of Alt vs. Ref read position bias"> ##INFO=<ID=SOR,Number=1,Type=Float,Description="Symmetric Odds Ratio of 2x2 contingency table to detect strand bias">
GATKCommandLineについて
この行には、ファイルを生成したツールで使用されたすべてのパラメータが含まれています。ここでは、GATK HaplotypeCallerで使用したコマンドラインを指します。 これらのパラメータには、ツールが受け付けたすべての引数と、適用された数値が含まれています。
##GATKCommandLine=<ID=GenomicsDBImport,CommandLine="GenomicsDBImport --genomicsdb-workspace-path gvcfs_db --variant ERR015377.g.vcf --variant ERR015425.g.vcf --variant ERR1035536.g.vcf --variant ERR1045266.g.vcf --variant ERR1106528.g.vcf --variant ERR1106529.g.vcf --variant ERR2000569.g.vcf --variant ERR211448.g.vcf --variant ERR343116.g.vcf --variant ERR405240.g.vcf --variant ERR405245.g.vcf --variant ERR450058.g.vcf --variant ERR450079.g.vcf --variant ERR484676.g.vcf --variant ERR562868.g.vcf --variant ERR580552.g.vcf --variant ERR636426.g.vcf --variant ERR636430.g.vcf --variant ERR701750.g.vcf --variant ERR701756.g.vcf --variant ERS032647.g.vcf --variant ERS032649.g.vcf --variant ERS347567.g.vcf --variant ERS347575.g.vcf --variant SRS399547.g.vcf --intervals sample.interval_list --reference /Hatena/GWAS/3D7_genomes/PlasmoDB-52_Pfalciparum3D7_Genome.fasta ・・・ ##GATKCommandLine=<ID=GenotypeGVCFs,CommandLine="GenotypeGVCFs --output merged.vcf --variant gendb://gvcfs_db --reference /Hatena/GWAS/3D7_genomes/PlasmoDB-52_Pfalciparum3D7_Genome.fasta --include-non-variant-sites false --merge-input-intervals false --input-is-somatic false --tumor-lod-to-emit 3.5 --allele-fraction-error 0.001 --keep-combined-raw-annotations false --use-posteriors-to-calculate-qual false --dont-use-dragstr-priors false --use-new-qual-calculator true --annotate-with-num-discovered-alleles false ・・・ ##GATKCommandLine=<ID=HaplotypeCaller,CommandLine="HaplotypeCaller --sample-ploidy 1 --emit-ref-confidence GVCF --output ERR015377.g.vcf --input ERR015377.markdup.bam --reference s /Hatena/GWAS/3D7_genomes/PlasmoDB-52_Pfalciparum3D7_Genome.fasta --use-posteriors-to-calculate-qual false --dont-use-dragstr-priors false --use-new-qual-calculator true --annotate-with-num-discovered-alleles false --heterozygosity 0.001 --indel-heterozygosity 1.25E-4 --heterozygosity-stdev 0.01 --standard-min-confidence-threshold-for-calling 30.0 --max-alternate-alleles 6 --max-genotype-count 1024 --num-reference-samples-if-no-call 0 --genotype-assignment-method USE_PLS_TO_ASSIGN ・・・
Contigについて
使用されたリファレンスアセンブリのコンティグ名、長さが記載されています。
##contig=<ID=Pf3D7_01_v3,length=640851> ##contig=<ID=Pf3D7_02_v3,length=947102> ##contig=<ID=Pf3D7_03_v3,length=1067971> ##contig=<ID=Pf3D7_04_v3,length=1200490> ##contig=<ID=Pf3D7_05_v3,length=1343557> ##contig=<ID=Pf3D7_06_v3,length=1418242> ##contig=<ID=Pf3D7_07_v3,length=1445207> ##contig=<ID=Pf3D7_08_v3,length=1472805> ##contig=<ID=Pf3D7_09_v3,length=1541735> ##contig=<ID=Pf3D7_10_v3,length=1687656> ##contig=<ID=Pf3D7_11_v3,length=2038340> ##contig=<ID=Pf3D7_12_v3,length=2271494> ##contig=<ID=Pf3D7_13_v3,length=2925236> ##contig=<ID=Pf3D7_14_v3,length=3291936> ##contig=<ID=Pf3D7_API_v3,length=34250> ##contig=<ID=Pf3D7_MIT_v3,length=5967>
3. バリアントレコードについて
各バリアントサイトのレコードは、次のように構成されています。
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT sample-nameが続く・・・ .
VCFレコードの最初の8列(INFOまで)は、バリアントサイトのレベルで測定されたスコアを表しています。
VCFファイルに複数のサンプルが含まれている場合、サイトレベルのアノテーションの中には、異なるサンプルからそのサイトについて得られた値の合計や平均値が含まれています。
遺伝子型や個々のサンプルレベルのアノテーション値などのサンプル固有の情報は、FORMAT列(9列目)およびsample-name列(10列目以降)に含まれています。ほとんどのプログラムでは、サンプル名のアルファベット順に並べていますが、必ずしもそうではありません。
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT ERR015377 ERR015425 ERR1035536 Pf3D7_01_v3 110 . A C 124.72 . AC=1;AF=0.071;AN=14;DP=99;FS=0.000;MLEAC=2;MLEAF=0.143;MQ=36.03;QD=31.18;SOR=3.258 GT:AD:DP:GQ:PL 0:9,0:9:99:0,135 0:2,0:2:49:0,49 .:0,0:0:.:0,0 Pf3D7_01_v3 260 . A G 251.07 . AC=2;AF=0.087;AN=23;BaseQRankSum=0.699;DP=188;FS=4.150;MLEAC=2;MLEAF=0.087;MQ=31.11;MQRankSum=0.301;QD=11.41;ReadPosRankSum=0.886;SOR=1.981 GT:AD:DP:GQ:PL 0:7,0:7:99:0,118 0:3,0:3:99:0,131 0:3,0:3:90:0,90 Pf3D7_01_v3 264 . C CTCTTACTTACTTACT 248.82 . AC=1;AF=0.048;AN=21;BaseQRankSum=-8.820e-01;DP=162;FS=6.990;MLEAC=1;MLEAF=0.048;MQ=36.03;MQRankSum=-1.400e-01;QD=24.88;ReadPosRankSum=-8.420e-01;SOR=4.407 GT:AD:DP:GQ:PL 0:7,0:7:99:0,118 0:3,0:3:99:0,131 0:3,0:3:90:0,90
お疲れ様でした。前半はここまでです。
後半は個別の項目について、詳しく見ていきましょう。
eupatho-bioinfomatics.hatenablog.com
よかったら他の記事もみていってください。 なお本記事は、大部分をGATK公式HPの内容を元に、自分で用意したデータを使って執筆しています。