複数のレファレンスゲノムへのマッピングでライブラリーDNAの組成を知る[FastQ screen]
FastQ screenの使い方
導入難易度 低い★☆☆☆☆高い
使用難易度 低い★☆☆☆☆高い
このツールで何ができる?
Fastqに対して、任意の数のリファレンスゲノムをマッピングしてリード数を比較することで、ライブラリーの含まれるリード数の組成を%で知ることができる。
私の使用感
一言で言うと、Fastq品質管理の個人的に必須な便利ツール。ベクターや大腸菌、ヒト由来のコンタミの確認を簡便・迅速に実行でき、一目でわかりやすい図を出力してくれます。その他、low complexityの判別やターゲット濃縮法で特定ゲノムを濃縮した際の濃縮効率の推定にも便利です。
Twitterで記事の更新をお知らせしているので、興味を持たれた方は是非フォローをお願いします。
フォローする @harrykun_blog
① 良好な品質の(コンタミ等のない)マウスサンプルの結果
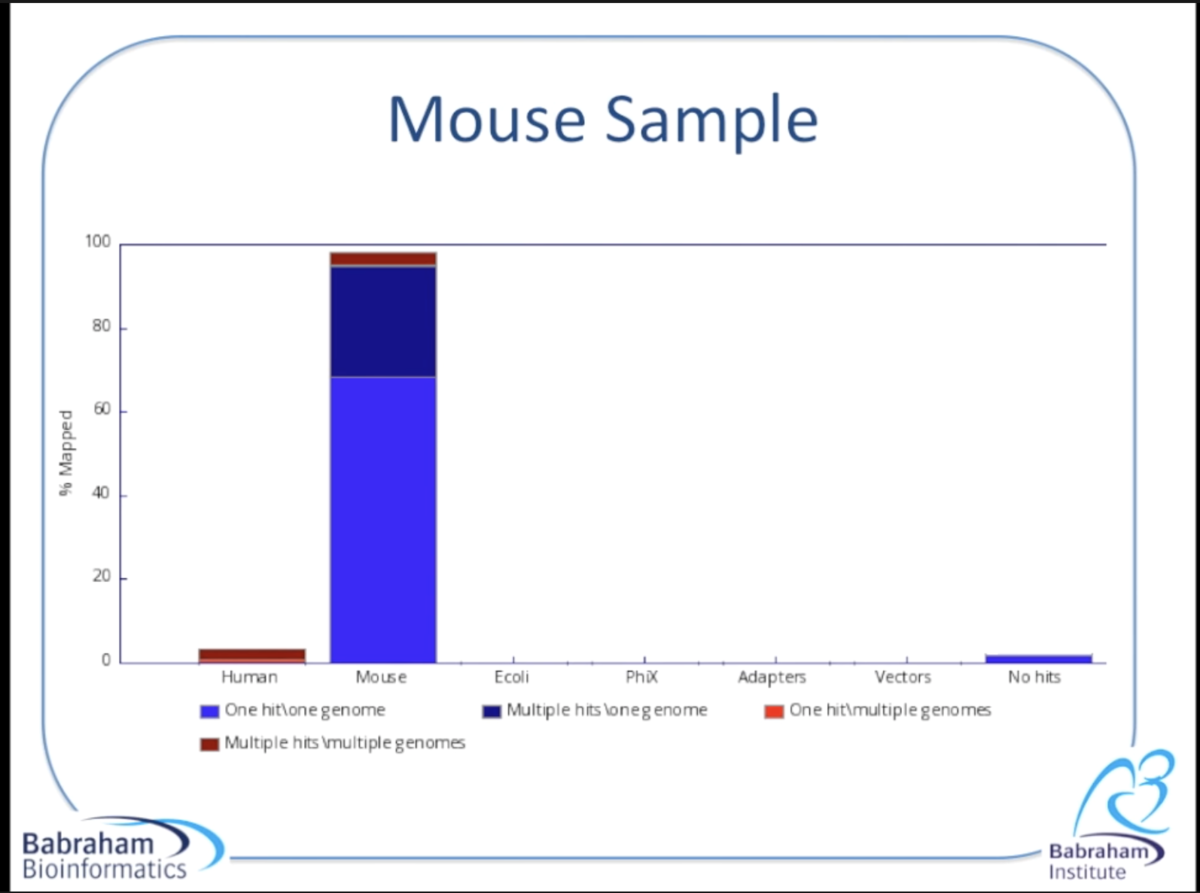 ② マウスゲノムのコンタミが見られる場合
② マウスゲノムのコンタミが見られる場合
 ③ サンプル調整に問題があるlow complexityな結果
③ サンプル調整に問題があるlow complexityな結果

上記の図表は作成者によるyoutube動画から引用した。
1. 1. FastQ screenのインストール
conda install -c bioconda fastq-screen
インストールが正しく導入されたか確認する。
fastq_screen
正しく導入されていれば、下記が表示される。
FastQ Screen - Map sequences against multiple genomes www.bioinformatics.babraham.ac.uk/projects/fastq_screen Contact: steven.wingett@babraham.ac.uk ・ ・ ・ --version Print the program version and exit.
eupatho-bioinfomatics.hatenablog.com
2. 必要なリファレンスゲノムの用意
マッピングしたい生物のリファレンスを入手する。 今回は、ヒトゲノム(hg38)をリンクからダウンロードした。 マラリア原虫のリファレンスの入手方法はこちら。 また、マラリア原虫と同属の原虫であるトキソプラズマに対しても同時にマッピングすることとして、ME49株のリファレンス配列を入手した。
なお、解析対象のfastqファイルには本ブログの過去記事↓
eupatho-bioinfomatics.hatenablog.com
で作成したマラリア原虫のfastq.gzファイルを例として用いる。
3. 構成ファイルの調整
fastQ-screenの解析には、あらかじめ情報を記した構成ファイルを用意する必要がある。
構成ファイルはこのページからsource code.tarをダウンロード・解凍する。
解凍されたフォルダの中にfastq_screen.conf.exampleというファイルがあるので、それを作業directoryにコピーする。
内容を確認してみると
<span style="color: #ff0000">cat fastq_screen.conf.example</span> # This is an example configuration file for FastQ Screen ############################ ## Bowtie, Bowtie 2 or BWA # ############################ ## If the Bowtie, Bowtie 2 or BWA binary is not in your PATH, you can set ## this value to tell the program where to find your chosen aligner. Uncomment ## the relevant line below and set the appropriate location. Please note, ## this path should INCLUDE the executable filename. #BOWTIE /usr/local/bin/bowtie/bowtie #BOWTIE2 /usr/local/bowtie2/bowtie2 #BWA /usr/local/bwa/bwa ############################################ ## Bismark (for bisulfite sequencing only) # ############################################ ## If the Bismark binary is not in your PATH then you can set this value to ## tell the program where to find it. Uncomment the line below and set the ## appropriate location. Please note, this path should INCLUDE the executable ## filename. #BISMARK /usr/local/bin/bismark/bismark ############ ## Threads # ############ ## Genome aligners can be made to run across multiple CPU cores to speed up ## searches. Set this value to the number of cores you want for mapping reads. THREADS 8 ############## ## DATABASES # ############## ## This section enables you to configure multiple genomes databases (aligner index ## files) to search against in your screen. For each genome you need to provide a ## database name (which can't contain spaces) and the location of the aligner index ## files. ## ## The path to the index files SHOULD INCLUDE THE BASENAME of the index, e.g: ## /data/public/Genomes/Human_Bowtie/GRCh37/Homo_sapiens.GRCh37 ## Thus, the index files (Homo_sapiens.GRCh37.1.bt2, Homo_sapiens.GRCh37.2.bt2, etc.) ## are found in a folder named 'GRCh37'. ## ## If, for example, the Bowtie, Bowtie2 and BWA indices of a given genome reside in ## the SAME FOLDER, a SINLGE path may be provided to ALL the of indices. The index ## used will be the one compatible with the chosen aligner (as specified using the ## --aligner flag). ## ## The entries shown below are only suggested examples, you can add as many DATABASE ## sections as required, and you can comment out or remove as many of the existing ## entries as desired. We suggest including genomes and sequences that may be sources ## of contamination either because they where run on your sequencer previously, or may ## have contaminated your sample during the library preparation step. ## ## Human - sequences available from ## ftp://ftp.ensembl.org/pub/current/fasta/homo_sapiens/dna/ #DATABASE Human /data/public/Genomes/Human_Bowtie/GRCh37/Homo_sapiens.GRCh37 ## ## Mouse - sequence available from ## ftp://ftp.ensembl.org/pub/current/fasta/mus_musculus/dna/ #DATABASE Mouse /data/public/Genomes/Mouse/NCBIM37/Mus_musculus.NCBIM37 ## ## Ecoli- sequence available from EMBL accession U00096.2 #DATABASE Ecoli /data/public/Genomes/Ecoli/Ecoli ## ## PhiX - sequence available from Refseq accession NC_001422.1 #DATABASE PhiX /data/public/Genomes/PhiX/phi_plus_SNPs ## ## Adapters - sequence derived from the FastQC contaminats file found at: www.bioinformatics.babraham.ac.uk/projects/fastqc #DATABASE Adapters /data/public/Genomes/Contaminants/Contaminants ## ## Vector - Sequence taken from the UniVec database ## http://www.ncbi.nlm.nih.gov/VecScreen/UniVec.html #DATABASE Vectors /data/public/Genomes/Vectors/Vectors
このうち、関連する項目だけを修正すればよい。
今回の場合、BWAを使用するので、BWAの正確なPATHを指定する。
Human、Plasmodium、ToxoplasmaのリファレンスもPATHを入力する。
また、自身のPCの性能に合わせて、THREADS の数値を変更する。
下記に変更した箇所だけを記載すると。
BWA /usr/local/bin/bwa THREADS 16 ## Human - sequences available from ## ftp://ftp.ensembl.org/pub/current/fasta/homo_sapiens/dna/ DATABASE Human /path/to/workingdir/Host_genomes/Homo_sapiens/NCBI/GRCh38/Sequence/BWAIndex/genome.fa ##Plasmodium_3D7 Sequence taken from PlasmoDB DATABASE Plasmodium /path/to/workingdir/3D7_genomes ##Toxoplasma_ME49 Sequence taken from ToxoDB-51 DATABASE Toxoplasma /path/to/workingdir/TgME49/ToxoDB-51_TgondiiME49_Genome.fasta
としたファイルをfastq_screen.confとして保存する。 これで準備は完了。あとは簡単です。
4. fastq_screenの実行解析
--aligner : マッピングツールを指定する。
--conf: 構成ファイルのPATHを指定する。
--outdir: 結果ファイルを入れる場所を指定する。
最後に全てのfastqファイルを入力する。
fasta='/path/to/workingdir/fastq' mkdir fastQscreen fastq_screen --aligner BWA --conf /path/to/workingdir/fastq_screen.conf --outdir /path/to/workingdir/fastQscreen\ $fastq/ERR1081237_1.fastq.gz \ $fastq/ERR1081238_1.fastq.gz\ $fastq/ERR1081239_1.fastq.gz\ $fastq/ERR1081241_1.fastq.gz\ $fastq/ERR1081242_1.fastq.gz\ $fastq/ERR1081254_1.fastq.gz\ $fastq/ERR1099214_1.fastq.gz\ $fastq/ERR1081255_1.fastq.gz\ $fastq/ERR1081257_1.fastq.gz\ $fastq/ERR1099215_1.fastq.gz\ $fastq/ERR1081261_1.fastq.gz\ $fastq/ERR1081262_1.fastq.gz\ $fastq/ERR1081263_1.fastq.gz\ $fastq/ERR1081264_1.fastq.gz\ $fastq/ERR1081265_1.fastq.gz\ $fastq/ERR1081283_1.fastq.gz\ $fastq/ERR1081284_1.fastq.gz\ $fastq/ERR1081285_1.fastq.gz\ $fastq/ERR1081287_1.fastq.gz\ $fastq/ERR1106549_1.fastq.gz\ $fastq/ERR1081237_2.fastq.gz\ $fastq/ERR1081238_2.fastq.gz\ $fastq/ERR1081239_2.fastq.gz\ $fastq/ERR1081241_2.fastq.gz\ $fastq/ERR1081242_2.fastq.gz\ $fastq/ERR1081254_2.fastq.gz\ $fastq/ERR1099214_2.fastq.gz\ $fastq/ERR1081255_2.fastq.gz\ $fastq/ERR1081257_2.fastq.gz\ $fastq/ERR1099215_2.fastq.gz\ $fastq/ERR1081261_2.fastq.gz\ $fastq/ERR1081262_2.fastq.gz\ $fastq/ERR1081263_2.fastq.gz\ $fastq/ERR1081264_2.fastq.gz\ $fastq/ERR1081265_2.fastq.gz\ $fastq/ERR1081283_2.fastq.gz\ $fastq/ERR1081284_2.fastq.gz\ $fastq/ERR1081285_2.fastq.gz\ $fastq/ERR1081287_2.fastq.gz\ $fastq/ERR1106549_2.fastq.gz

4. 解析結果
解析が終わると結果がhtmlとtxtで出力される。
一つ一つ結果を見ることもできますが、今回はmultiqcを使って一つにまとめた。
multiqcについては後日紹介する。
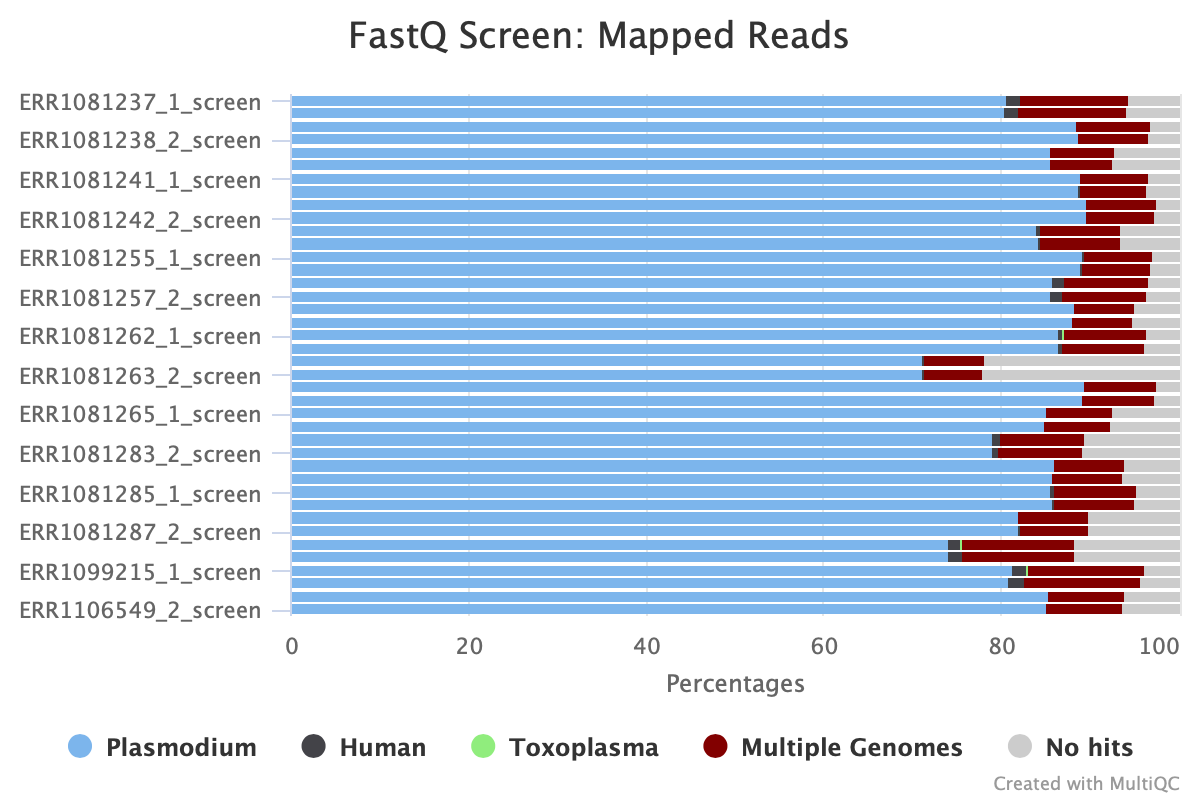
結果から、少しNo hitが多いような気がしますが、少なくともヒトゲノムのコンタミがないことは分かりました。 目的に応じてマッピングする生物を選んで使ってみてください。
今回はこれで終わりです。 よければ他の記事のも見ていってください。